AHR binding affinity
Endpoint
Aryl hydrocarbon receptor (AhR) modulates the biochemical and toxic effects of a wide variety of environmental compounds and plays an important role in adaptation of organisms to environmental stress [1]. Responses mediated by AhR include transcriptional induction of Phase I and Phase II metabolism genes such as CYP1A1 [2].
Data
Training set of the AhR model consists of 142 organic compounds collected from different literature sources. Covered are four typical classes of AhR ligands: polychlorinated biphenyls, polychlorinated dibenzofurans, polychlorinated dibenzodioxins, ellipticines and flavones. Relative equivalent potencies (REP) of these substances towards AhR have been estimated:
|
|
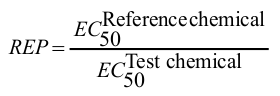
where EC50 is the half maximal effective concentration. Reference chemical is the most potent AhR agonist 2,3,7,8-tetrachlotrodibenzo-p-dioxin.
Another training set consisting of 51 chemicals with gene expression (GE) data have also been collected. Of them, 23 chemicals have data for both AhR binding and GE. These chemicals were used to establish a relation between binding and GE.
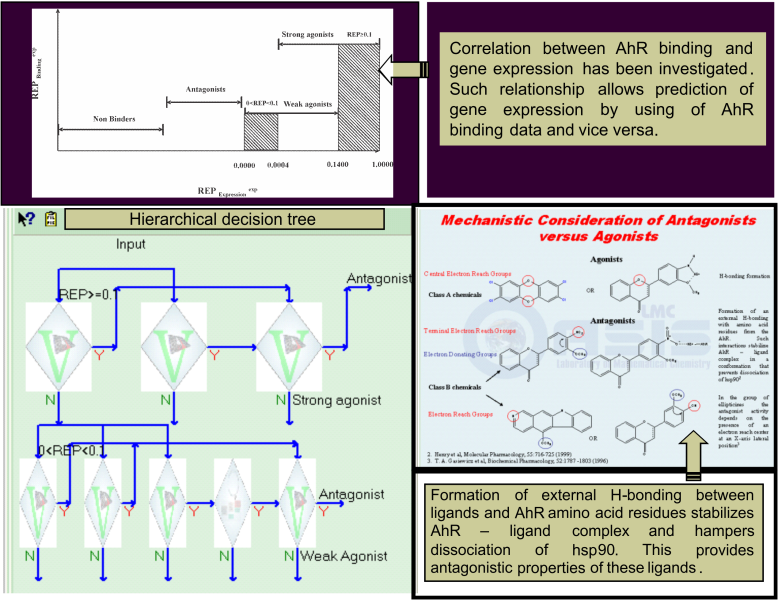
The TIMES AhR model has been developed for predicting: AhR binding, agonistic/antagonistic properties and gene expression.
AhR binding
A battery of structure-activity relationship (SAR) models for predicting AhR binding based on two distinct mechanisms has been developed [3]. One of these mechanisms is based on electron charge transfer from ligands to the Ah receptor. This mechanism is associated with dioxin-like compounds, where a favorable interaction with a receptor nucleophilic site in the central part of the ligand and with electrophilic sites at both sides of the principal molecular axis is required. Same mechanism is also associated with polycyclic aromatic hydrocarbon (PAHs), where a stacking-type of interaction with the AhR is required. The second AhR binding mechanism is associated with biphenyl-like compounds which unlike the above chemical classes are found to accommodate electron density from the receptor.
These two mechanisms are studied across three activity ranges: strong binders with REP ≥ 0.1 (30 chemicals), weak binders with 0 < REP < 0.1 (52 chemicals), and non-binders with REP = 0 (60 chemicals). Structure of the AhR binding model is presented in Figure 1:
Figure 1. Structure of the AhR receptor binding model
Individual SAR models have been developed for dioxin-like compounds, biphenyls and PAHs in each activity bin. SARs consist of structural and parametric boundaries. First, minimum structural requirements for interacting with AhR are developed. Second, additional parametric boundaries have been added for completing definition of the model. While minimum structural requirements could bring binding and non-binding effect, the additional parametric boundaries provide the sufficient conditions for binding affinity only.
First, when a new chemical is submitted for prediction, its AhR binding affinity is estimated by application of the individual SARs in the high activity bin. If the complete set of boundaries are met, the substance is assigned to be strong AhR binder. For a negative responce, the substance is submitted to the SAR models associated with low activity bin. Meeting the criteria for activity in this range classifies the substance to be week AhR binder.
Agonism/antagonism
The model is also trained to identify agonists and antagonists. While agonistic properties are related to ability of substances to elicit gene expression when bind to a receptor, antagonism is obtained when substances fail to trigger gene expression. In case of antagonism, chemicals bind to the receptor surface through the electron-donating properties of electron-rich groups which retain the receptor in the cytosol and does not allow AhR-dependent signal transduction [4]. Hence, SAR model accounting for antagonism has been derived and incorporated in the model, as illustrated in Figure 2:
Figure 2. Screening for antagonism
First, strong binders are screened for antagonistic properties. If criteria for antagonism are met, substances are assigned to be antagonist. Negative response in this SAR classifies substances to be strong agonist. Similarly, weak binders could be classified as antagonists or weak agonists.
Gene expression
Categorization of AhR binders as agonists or antagonists is found to correlate with their gene expression. The highest increase in gene expression was elicited by strong agonists, followed by weak agonists producing lower increases in gene expression, whereas all antagonists (and non-AhR binders) were found to have no effect on gene expression. This relationship helps predicting AhR binding affinity using only gene expression data which are more frequently available in literature.
Performance
For the training set of 142 chemicals, the AhR binding model was able to predict correctly 83% of the strong binders, 73% of the weak binders and 63% of the non-binders, i.e., an overall performance of 71% is achieved.
Domain
Applicability domain of the AhR model consists of two sub-domain layers: general parametric requirements and structural features [5]. Two chemical subsets are used for deriving the model domains. The first subset includes the training set chemicals which are correctly predicted by the models, whereas the second subset comprises training set chemicals which are incorrectly predicted by the models.
The correct chemical subset is used for defining the general parametric requirements. Extracted are specific ranges of the molecular weight (MW) and the 1-octanol/water partition coefficient (log KOW):
- Molecular weight MW (in Da) ϵ [178; 578],
- log KOW ϵ [2; 9].
The atom-centered fragments extracted from the correct subset of chemicals are used to define the structural domain. Briefly, the structural domain is assessed based on atom-centered fragments, extracted from correctly and incorrectly predicted (i.e., false positives and false negatives) substances from the model training sets by accounting for the atom type, hybridization and attached H-atoms of the central atom and its first neighbours. If the neighbour is a heteroatom then the diameter of the fragment is increased up to three consecutive heteroatoms or to the first carbon atoms in sp3 hybridization. In order to assess if a new chemical belongs to the structural domain, the system partitions the chemical to atom-centered fragments, which are then matched to the fragments extracted from the correct and incorrect chemical subsets. The new chemical is estimated to belong to the structural domain only when its atom-centered fragments are found in the list of correct fragments.
Reporting
Predictions from the AhR binding model could be reported as a tab delimited text file providing the following information for: chemical identity (CAS number, Name, SMILES), observed binding affinity (ranges), predicted binding affinity (ranges), binding mechanism, applicability domain, etc.
.
References
- Hoffman, E.C., Reyes, H., Chu, F.F., Sander, F., Conley, L.H., Brooks, B.A., and Hankinson, O. 1991. Cloning of a factor required for activity of the Ah (dioxin) receptor, Science 252, pp. 954-958.
- Denison, M.S., Fisher, J.M., Whitlock, J.P. 1988. The DNA recognition site for the dioxin-Ah receptor complex. Nucleotide sequence and functional analysis, J. Biol. Chem. 263, pp. 17221-17224.
- Petkov, P.I., Rowlands, J.C., Budinsky, R., Zhao, B., Denison M.S., Mekenyan, O. 2010. Mechanism-based common reactivity pattern (COREPA) modelling of aryl hydrocarbon receptor binding affinity, SAR and QSAR in Environmental Research, Vol. 21, pp. 187-214.
- Henry, E.C., Kende, A.S., Rucci, G., Totleben, M.J. 1999. Flavone Antagonists Bind Competitively with 2,3,7,8-Tetrachlorodibenzo-p-Dioxin to the Aryl Hydrocarbon Receptor But Inhibit Nuclear Uptake and Transformation, Mol. Pharmacol. 55, pp. 716-725.
- Dimitrov, S., Dimitrova, G., Pavlov, T., Dimitrova, N., Patlewicz, G., Niemela J., Mekenyan, O. 2005. J. Chem. Inf. Model., Vol. 45, pp. 839-849.
49.
AHR Binding
Model Features
Click the image for a larger view