Androgen binding affinity
Endpoint
The presence of so-called endocrine disruptors in the environment has become a worldwide environmental concern. It has been concluded that such compounds can act in a same way as endogenous hormones eliciting a variety of adverse effects in both humans and wildlife.
The concern associated with xenobiotics that may bind to the androgen receptor has created a need for development of model to predict their possible effect. The androgen receptor-binding model assesses the in vitro relative binding affinity (RBA) of chemicals to interact with the androgen receptor (AR).
Data
A structurally diverse training data set containing 202 chemicals was obtained from the National Center for Toxicology Research [1]. The binding affinities of the chemicals from the training set were obtained from a competitive radiometric binding assay, using radiolabeled [3H]-R1881 as the tracer and androgen receptor recombinant rat protein expressed in Eshcerichia coli. The relative binding affinity for each chemical was determined as a ratio between the 50% inhibition of [3H]R1881 (IC50) and the IC50 of the competitor.
Model
Chemicals in the training set were categorized according to their potency and grouped into four activity bins: highly active with RBA>10%; moderate with 0.1%<RBA<10%; low with 0.001%<RBA<0.1% and non active with RBA<0.001%. For each potency bin, the ability of the chemicals to bind the receptor was related to the distances between nucleophilic sites and structural features describing electronic and hydrophobic interactions between the receptor and ligands. Categorical models were derived for each binding affinity range in terms of specific distances, local (maximal donor delocalizability associated with the oxygen atom), global nucleophilicity (partial positive surface areas and energy of the highest occupied molecular orbital) and hydrophobicity (log Kow) of the molecules. An integral screening tool for predicting binding affinity to AR was constructed as a battery of models each associated with different activity bins [2] (Figure 1).
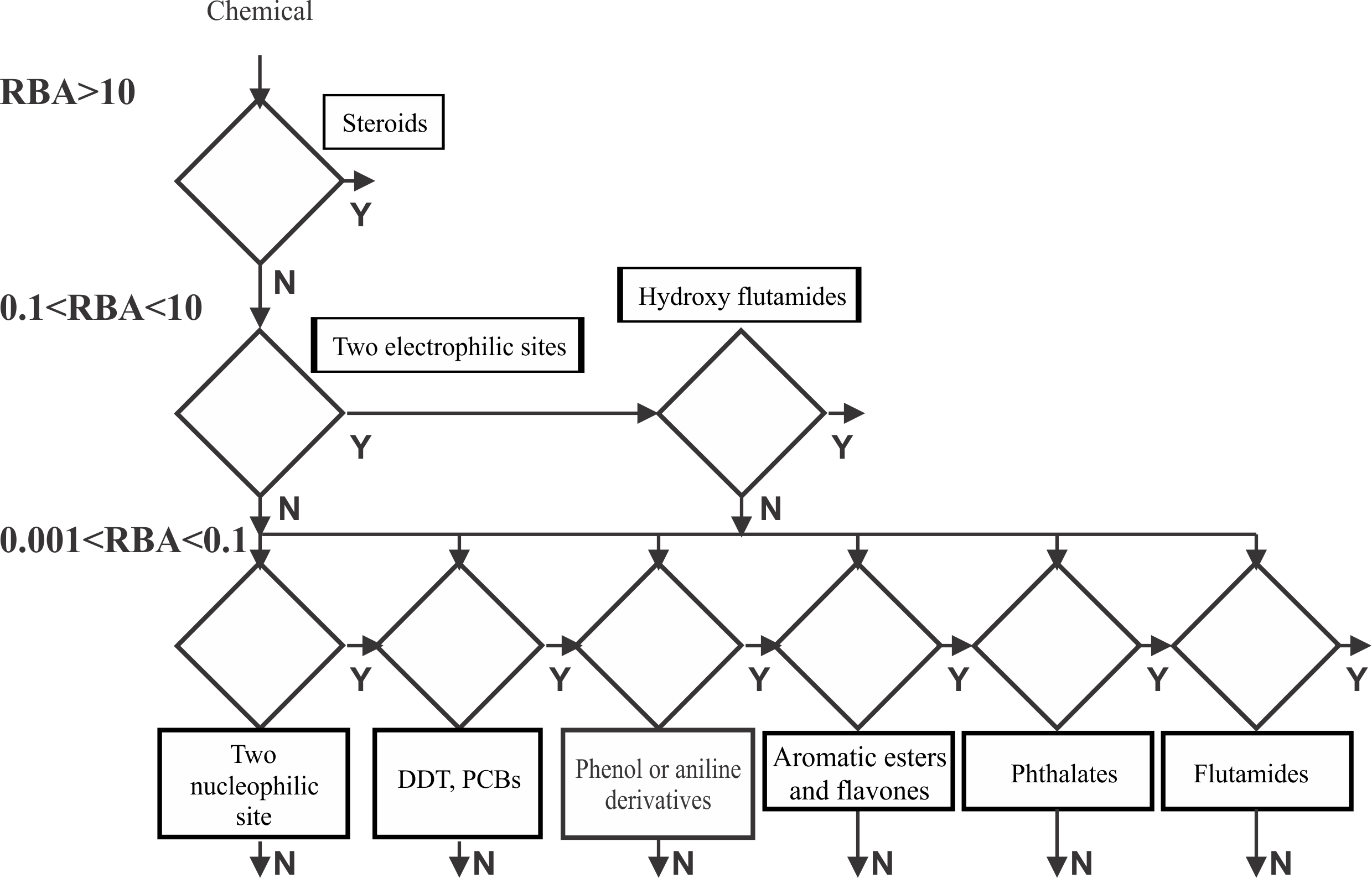
Figure 1. Structure of the AR binding model model
When a chemical is submitted for prediction the requirements for strong AR binding are first applied. If the chemical does not respond to all of them, then the requirements for the lower activity bins are applied sequentially. If the chemical passes through the activity bins without meeting a binding requirement, then the ultimate prediction is not AR binder.
Performance of the АR binding modelDomain
According to the performance of the model when applied on the training set chemicals (140 binders and 62 non binders), correct predictions are provided for:
101 out of 132 chemicals ER binders as parents (Sensitivity = 78 %)
54 out of 59 chemicals non binders as parents (Specificity = 92%)
*Predictions for all chemicals which do not exceed model probability threshold are excluded
Domain
Applicability domain of the ER binding model consists of four sub-domain layers: general parametric requirements, structural features and interpolation space [4]. Two chemical subsets are used for deriving the model domains. The first subset includes the training set chemicals which are correctly predicted by the models, whereas the second subset comprises training set chemicals which are incorrectly predicted by the models.
The correct chemical subset is used for defining the general parametric requirements. Extracted are specific ranges of the molecular weight (MW) and the 1-octanol/water partition coefficient (log KOW):
• Molecular weight MW (in Da) ϵ [90; 550],
• log KOW ϵ [-0.7; 10].
The atom-centered fragments extracted from the correct subset of chemicals are used to define the structural domain. Briefly, the structural domain is assessed based on atom-centered fragments, extracted from correctly and incorrectly predicted (i.e., false positives and false negatives) substances from the model training sets by accounting for the atom type, hybridization and attached H-atoms of the central atom and its first neighbours. If the neighbour is a heteroatom then the diameter of the fragment is increased up to three consecutive heteroatoms or to the first carbon atoms in sp3 hybridization. In order to assess if a new chemical belongs to the structural domain, the system partitions the chemical to atom-centered fragments, which are then matched to the fragments extracted from the correct and incorrect chemical subsets. The new chemical is estimated to belong to the structural domain only when its atom-centered fragments are found in the list of correct fragments.
Interpolation space - estimates the population density of the parametric space defined by the explanatory variables of the QSAR models by making use the training set chemicals.
Reporting
Predictions from the AR binding model could be reported as a tab delimited text file providing the following information for: chemical identity (CAS number, Name, SMILES), observed binding affinity (ranges), predicted binding affinity (ranges), binding mechanism, applicability domain, etc.
External validation of the AR binding model
The performances of the model have been externally validated by screening exercise performed by the Dow Chemical Company [6]. In this study a set of more than 1800 ToxCast™ Phase II compounds were used for evaluation of the sensitivity and specificity of the model. The sensitivity (correct predictions for binders) was found to be greater than 90% and specificity (correct predictions for non-binders) 80% for predictions in the model domain. The authors recommend the use of the model as reliable tool for identification of compounds able to bind the androgen receptor.
Refrences
- R. Serafimova, M. Todorov, D. Nedelcheva, T. Pavlov, Y. Akahori, M. Nakai, and O. Mekenyan. QSAR and mechanistic interpretation of estrogen receptor binding, SAR QSAR Environ. Res. 18, (2007), pp. 1-33
- J. Katzenellenbogen, Effectiveness of QSAR for prescreening of endocrine disruptor hazard, SCOPE/IUPAC International Symposium on Endocrine Active Substances, Yokohama, Japan, 2002.
- O. Mekenyan and R. Serafimova. Mechanism-Based Modeling of Estrogen Receptor Binding Affinity A COREPA Implementation. CRC Press, (2009), pp. 259-293
- S. Dimitrov, G. Dimitrova, T. Pavlov, N. Dimitrova, G. Patlevisz, J. Niemela and O. Mekenyan, J. Chem. Inf. Model. 45 (2005), pp. 839-849.
- Gaido, K.W., Leonard, L.S., Maness, S.C., Hall, J.M., McDonnell, D.P., Saville, B & Safe, S. (1999). Differential interaction of the methoxychlor metabolite 2,2-Bis-(phydroxyphenyl)- 1,1,1-trichloroethane with estrogen receptors α and β. Endocrinology 140, 5746-5753.
- B. Bhhatarai, D. M. Wilson, P. S. Price, S. Marty, A. K. Parks, and E. Carney Evaluation of OASIS QSAR Models Using ToxCast™ in Vitro Estrogen and Androgen Receptor Binding Data and Application in an Integrated Endocrine Screening Approach.. Environ Health Perspect. 2016, 12, 9, 1453-1463
Androgen binding
Model Features
Click the image for a larger view
